Electrosynthesis Awakens Dormant Bonds
Organic electrosynthesis uses electricity to precisely guide bond formation and selectivity, enabling green, controllable molecular design.
Sponsored by

Sponsored by
Organic electrosynthesis is the modern conductor who directs molecules not with heat, not with acids or bases, but with a stream of electricity.
As electrons flow gently across the electrode surface, they apply a soft yet precise electric-field nudge to molecules, causing previously inert chemical bonds to cleave and reorganize at just the right moment, composing entirely new molecular architectures.
The key advantage of electrosynthesis lies in its ability to precisely control reaction patterns, selectivity, and even stereochemistry simply by adjusting external knobs such as current, electrode material, and electrolyte. This green transformation method is reshaping the boundaries of modern synthetic organic chemistry.
Recently, Associate Professor Chen Zhu from the College of Science at the Eastern Institute of Technology, Ningbo, and his collaborators have provided new insights into this field. They have achieved consecutive significant breakthroughs in photoelectro-cooperative catalysis and precise electrochemical regulation. The related research findings have been published consecutively in top-tier chemistry journals Nature Synthesis, Nature Chemistry, and Nature Communications.
1. Electrochemical Switch System: Precisely Determining the Pathway for Arene Alkylation
Imagine having a chemical universal toolbox containing a versatile molecular feedstock (like common benzoic acid). You wish to use this feedstock to produce two completely different, yet highly valuable, pharmaceutical intermediates. Traditionally, this would require two entirely separate sets of apparatus, reagents, and procedures. Now, scientists have developed an ingenious electrochemical smart switch. Simply by making two straightforward swaps—like changing a battery and a seasoning—within the same reaction electrochemical pot, and then applying a mild electric current under gentle conditions, the transformation occurs.
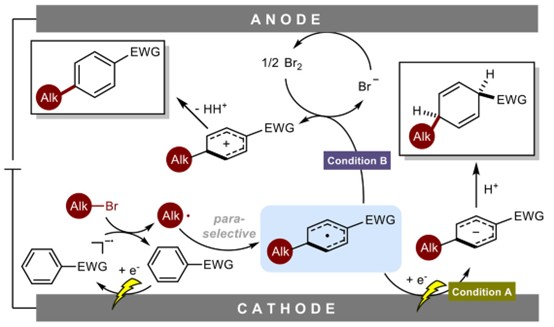
In collaboration with Professor Wujiong Xia and Professor Lin Guo's team from Harbin Institute of Technology (Shenzhen), Professor Chen Zhu's team developed a remarkable electrochemical switch system: merely by swapping the anode (metal Zn ↔ Nb) and the electrolyte (BF₄⁻↔ Br⁻), the same reaction setup can be directed towards two entirely distinct transformations. This research is akin to crafting a master key and a smart switch. Using the same electrochemical reactor, switching the electrode and electrolyte allows one to select different programs, guiding the same starting molecule down two divergent, valuable synthetic pathways.
This is not merely an innovation of the classic Birch reaction; it also opens a concise, green, and highly controllable new route for the precise late-stage functionalization of complex molecules. The related research was published in Nature Chemistry on November 17.
Two Selective Pathways for Arene Alkylation Reaction | Image provided by the research team
Pathway A: Zn Anode + BF₄⁻→ syn-1,4-Hydroalkylation (Dearomatization)
Generates structurally intricate syn-1,4-cyclohexadienes (equivalent to a greener, milder upgraded Birch reduction).
Pathway B: Nb Anode + High [Br⁻] → para-Selective C(sp²)–H Alkylation
Achieves highly selective C–H alkylation directly at the para-position of the arene ring.
The entire reaction system operates without any transition-metal catalysts under mild conditions (0 °C, 5 mA) and is amenable to gram-scale synthesis and flow electrolysis, offering a novel approach for the late-stage modification of drug molecules such as steroids, ibuprofen, and citronellol. This represents a significant innovation over traditional Birch reactions and remote C–H functionalization.
Link: https://doi.org/10.1038/s41557-025-02001-9
2. AC Photoelectrocatalysis: One-Step Transformation of Simple Alcohols into Highly Stereoselective Chiral Molecules
If researchers aim to modify a complex drug molecule—for instance, the common anti-allergy drug loratadine—by precisely excising a small fragment from its molecular backbone and installing a new moiety with a specific spatial orientation to obtain a novel chiral drug molecule, traditional methods often require multiple steps under harsh conditions with compromised precision. Professor Chen Zhu's team found a more ingenious approach: using LED light combined with controlled electric current to accomplish this challenging transformation at room temperature.
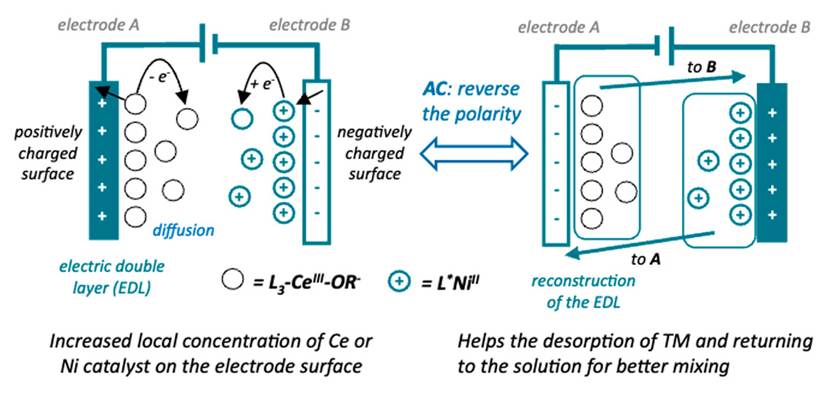
Mechanism of AC Photoelectro-Catalysis | Image provided by the research team
The core innovations of this system include:
LMCT (Ligand-to-Metal Charge Transfer) photocatalysis to generate radicals.
Chiral Nickel catalysis enabling asymmetric bond construction.
AC waveform restructuring of the electrical double layer to prevent catalyst adsorption and deactivation, boosting efficiency from 20% (DC) to 86% (AC).
Professor Chen Zhu's team introduced alternating current (AC) into a photoelectrochemical asymmetric catalytic (AC-PEC) system for the first time. Their theoretical calculations also elucidated a novel mechanism involving concerted cleavage of the Ce–O bond and the β-C–C bond within the cerium catalytic cycle, deepening the understanding of how light and electricity cooperatively direct molecular behavior. The related research was published in Nature Synthesis on September 29.
This method is compatible with various complex drug skeletons and has been applied to the late-stage chiral modification of drug molecules like loratadine, propranolol, and fluoxetine. DFT calculations revealed the unique mechanism of concerted cleavage of the Ce–O and the β-C–C bond in the cerium cycle, providing fresh insight into photoelectro-cooperative catalysis.
Link: https://doi.org/10.1038/s44160-025-00875-8
3. A Remarkable Switch: Unlocking a Dual-Mode Decyanation/Benzylic C–H Germylation Reaction
In collaboration with Professor Magnus Rueping's team at King Abdullah University of Science and Technology (KAUST), Professor Chen Zhu's team discovered a remarkable switch. Simply by adjusting whether cobalt bromide is added, completely different reaction pathways can be selected within the electrolysis system. The related research was published in Nature Communications on August 6.
With Co-catalyst: Alkyl Nitrile → Decyanative Germylation (Forming C–Ge Bond)
Achieved, for the first time, the electrocatalytic decyanative germylation of various primary, secondary, and tertiary alkyl nitriles.
Without Co-catalyst: Benzylic Nitrile → Direct Benzylic C–H Germylation
Selectively abstracts the α-C–H hydrogen, enabling metal-free, precise germylation.
The reaction conditions are simple (room temperature, 4 mA, undivided cell), using inexpensive and readily available Me₃GeCl as a general germanium source, with broad substrate compatibility.
Mechanistic studies indicate:
Cobalt pathway: Co(I)/Co(III) cycle + decyanated radical → coupling.
Cobalt-free pathway: Cathodically generated germanium radicals → site-specific hydrogen atom transfer.
This is like providing chemists with a new, intelligent set of molecular Lego tools. Through a simple conditional switch, one can selectively construct structurally diverse organogermanium molecules from the same class of starting materials. These germanium-containing molecules hold unique potential in fields like semiconductor materials and drug discovery. This method offers a new practical tool for the green and efficient synthesis of complex organogermanium compounds.